特殊检测类型:生物发光法,bio-luminescence
1. 主要涉及ATP类的试剂盒,消耗能量ATP,发光(萤火虫)。
2. 主要的实验:荧光素酶分析实验。
使用仪器:
1. 化学发光仪,
2. 光度计(Luminometer),
3. 多功能酶标仪(含化学发光,生物发光检测模块)。
ELISA实验数据不稳定,背景高:洗涤很关键
1.试剂盒使用前,通常需要在室温下平衡30-60min。(温度对酶催化反应有影响)。
2.洗板时需保证微孔板平放,将洗涤液注满各孔,但尽量避免漏液溢液,以每孔300 µl为宜;板洗次数一般为3次,要保证洗板的浸泡时间,每次浸泡时间一般为30-60 秒;尽量减少洗液残留量,推荐每次洗板后进行拍板,并及时更换吸水纸,避免吸水纸的碎屑粘到反应孔内。
用什么96孔板?

96孔板:Solid plate & Strip(条) plate

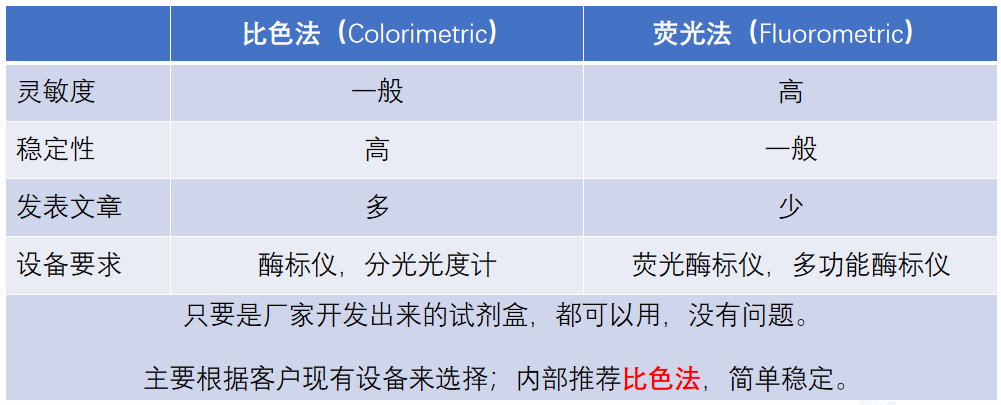
比色法 & 荧光法:哪个更好?
比色法:特定波长光穿透样品,光被吸收的情况。(酶标仪)(分光光度计)
荧光法:特定波长光,激发染料,发出其它波长光的情况。(荧光/多功能,酶标仪)
如何确认同一个蛋白(产品替换)
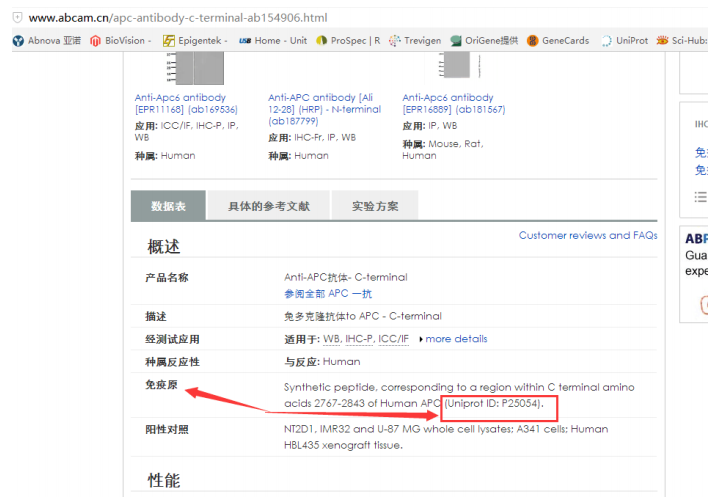
1.找到这个抗体的免疫原,什么蛋白的抗体。【留意应用类型和反应种属】 或者,在【靶标】这一栏观察,SwissProt 这一栏,至于是选人的还是小鼠的, 根据客户实验需求来。可以点击进去,进入 Uniprot 数据库。
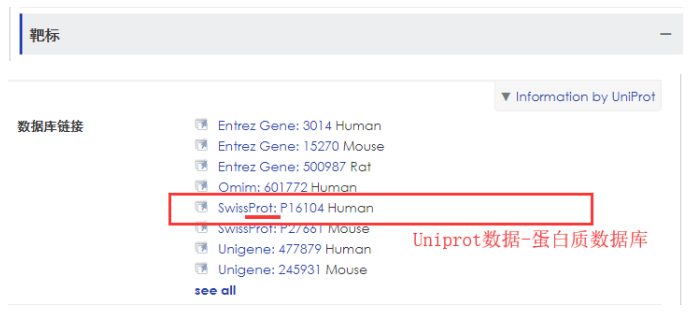
2. (Uniprot ID: P25054).这是数据库的 ID,类似身份证,打开数据库:http://www.uniprot.org/ ,输入 ID,P25054,搜索。
3.
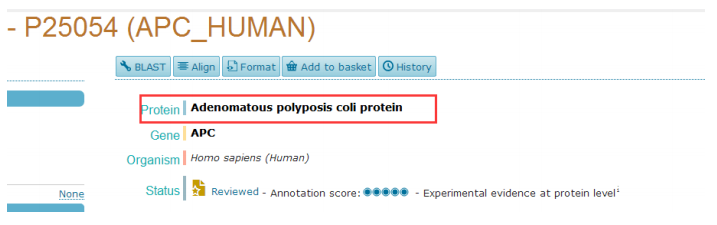
全名如图,可以用全名和缩写去搜索抗体。并在其它品牌的产品页面,留意观 察抗原的信息,是否与数据库的一致 ···这个 ID 相同,就像身份证号码一样,指示是同一个人。
案例:(问题,是否为同一个蛋白,是否可以替换为 Abbkine 产品)
1. http://www.alomone.com/p/anti-angiotensin-(1-7)_mas_receptor/agp-099/119
2. http://www.abbkine.com/product/mas1-polyclonal-antibody-abp57206/
其它现象:
●膜上多处出现黑点或黑斑原因:抗体与封闭试剂发生非特异性的结合。
●反白(条带显白色)原因:目的蛋白含量太高或者一抗浓度偏高。
●蛋白分子量偏低或偏高原因:胶浓度不适合,高分子量要用低浓度胶;小分子蛋白要用高浓度胶。
Westernblot 结果中无信号或显示信号弱
可能的原因及建议:
●检测样本不表达目的蛋白选择表达量高的细胞作为阳性对照,用于确定检测样本是否为阴性。
●检测样本低表达目的蛋白提高上样量,裂解液中注意加入蛋白酶抑制剂。
●转移不完全或过转移可以用丽春红染膜并结合染胶(考马斯亮蓝)后确定条带是否转至膜上或转移过 头;适当调整转膜的时间和电流。
●抗体不能识别测试种属的相关蛋白购买抗体前应当认真阅读抗体说明书,确定其是否能够交叉识别测 试种属的对应蛋白。
●一抗孵育时间不足建议 4℃结合过夜。
●二抗与一抗不匹配 选择针对一抗来源的种属的抗体。
●洗膜过度洗膜时间不宜过长,加入的去垢剂不宜过强或过多,建议使用0.1%的弱去垢剂 Tween-20。
安全问题:
操作有毒试剂时,带手套和口罩,且操作挥发性试剂应在通风橱中进行。
Westernblot 结果中杂带较多
可能的原因及建议:
●目的蛋白有多个修饰位点(磷酸化位点、糖基化位点、乙酰化位点等),本身可以呈现多条 带。查阅文献或进行生物信息学分析,获得蛋白序列的修饰位点信息,通过去修饰确定蛋白实际大小。
●目的蛋白有其它剪切本查阅文献或生物信息学分析可能性。
●样本处理过程中目的蛋白发生降解加入蛋白 酶抑制剂;样本处理时在冰上操作。
●上样量过高,太敏感适当减少上样量。
●一抗特异性不高重新选择或制备 高特异性的抗体。
●一抗不纯纯化抗体
●一抗或者二抗浓度偏高降低抗体浓度。
电泳中常出现的一些现象:
●︶条带呈笑脸状 原因:凝胶不均匀冷却,中间冷却不好;电泳系统温度偏高。
●︵条带呈皱眉状原因:可能是由于装置不合适,特别可能是凝胶和玻璃挡板底部有气泡,或者两边聚 合不完全。
●拖尾原因:样品溶解不好。
●纹理(纵向条纹)原因:样品中含有不溶性颗粒。
●条带偏斜原因:电极不平衡或者加样 位置偏斜。
●条带两边扩散原因:加样量过多。
Westernblot 结果中背景较高
可能的原因及建议:
●膜封闭不够延长封闭的时间;选择更加适合的封闭液。
●一抗稀释度不适宜对抗体进行滴度测试,选择最适宜的抗体稀释度。
●一抗孵育的温度偏高,建议 4℃结合过夜。
●选择的膜容易产生高背景,一般硝酸纤维素膜的背景会比PVDF 膜低。
●膜在实验过程中干过实验过程中要注意保持膜的湿润。
●检测时曝光时间过长,减少曝光时间。
产品订购:sales@amyjet.com
邮政编码:430070
公司地址:武汉市洪山区光谷大道35号
光谷总部国际二期时代1栋13楼
提示:本公司所有产品仅供科研使用,不用于临床诊断。
版权所有:艾美捷科技有限公司 鄂ICP备10204150号-1 鄂公网安备:42018502004523号
第二类医疗器械经营备案凭证:鄂汉药监械经营备20234324号

微信扫码在线客服